The Orphan Drug Development Guidebook (ODDG)
The Orphan Drug Development Guidebook is a patient focused guidebook that describes the available tools, incentives, resources and practices specific for developing traditional and innovative drugs/therapies for rare disease indications and how to best use them. It can be used by academic, non-profit organizations, small and larger (innovative) biotechs and patient-driven drug developers. The Guidebook currently has two chapters: the chapter dedicated to orphan drug development (ODDG) focused on small molecules or innovative therapies, and a chapter dedicated to drug repurposing (DRG).
More information can also be found in the papers published on the Orphan Drug Development Guidebook, the Drug Repurposing Guidebook and START checklist.
Key Drug Development Milestones
Explore the guide
LET'S START!
Orphan Drug Development Cases
TRADITIONAL TECHNOLOGY TARGETING A 'SUFFICIENTLY WELL UNDERSTOOD' RARE DISEASE
Basic case of development of a traditional, well-understood pharmaceutical (such as a small molecule or a protein) targeting a 'sufficiently well understood' rare disease to be registered in EU, US and Japan
-
Assumptions
- The drug is a well-understood pharmaceutical product
- There is already a non-negligible body of knowledge around the disease in the regulatory and medical community
- Patient population is sufficiently understood and non-clinical development is possible (relevant animal model exists)
- Disease is present in children and adults
- Funding the development is not a problem, but return of investment might be
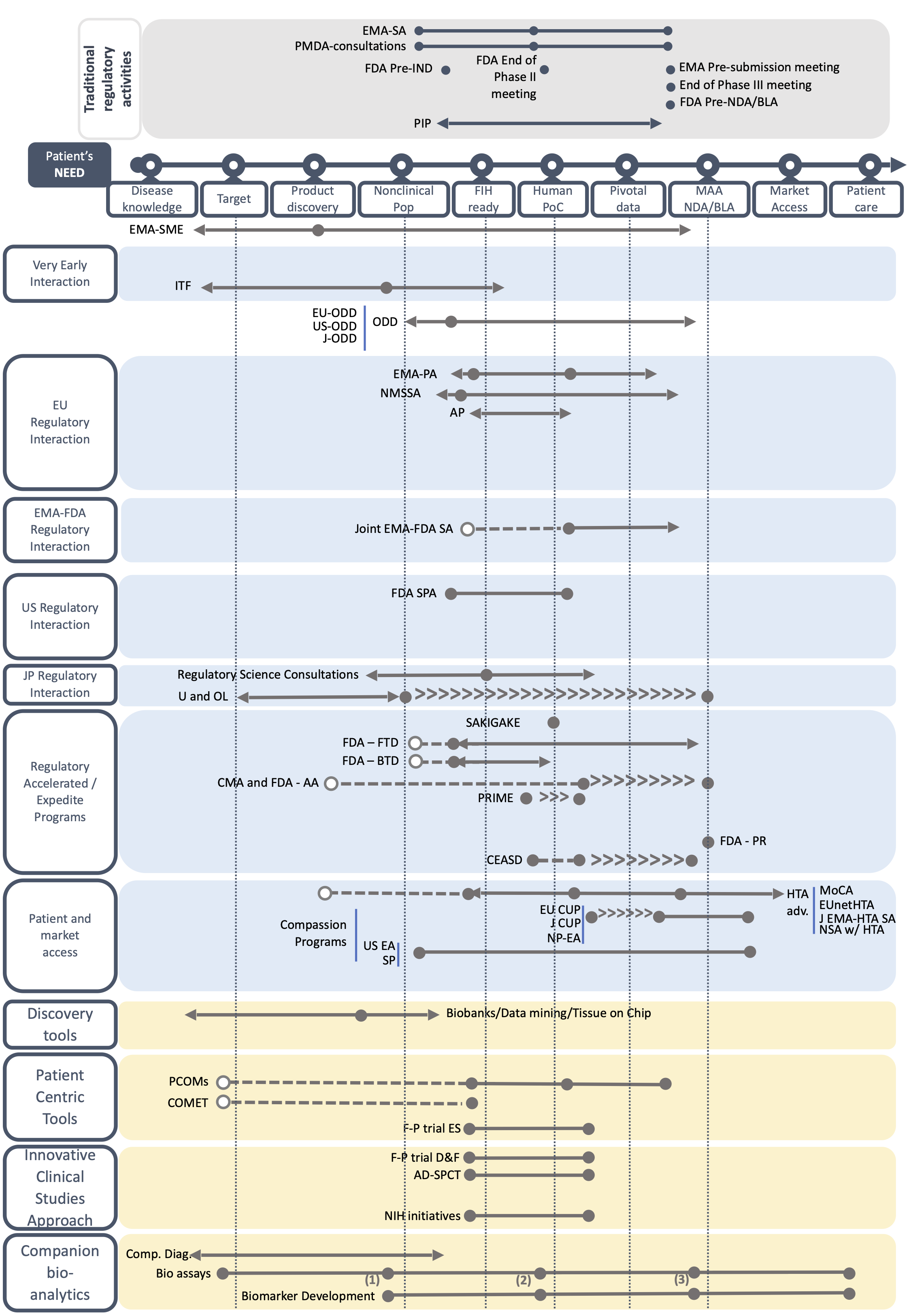
HIGHLY INNOVATIVE TECHNOLOGY TARGETING A 'SUFFICIENTLY WELL UNDERSTOOD' RARE DISEASE
Case of development of a highly innovative technology (i.e. advanced therapy medicinal products) targeting a 'sufficiently well understood' rare disease to be registered in EU, US and Japan
-
Assumptions
- The drug is a highly innovative technology like the advanced therapy medicinal products
- There is already a non-negligible body of knowledge around the disease in the regulatory and medical community
- Patient population is sufficiently understood and non-clinical development is possible (relevant animal model exists)
- Disease is present in children and adults
- Funding the development is not a problem, but return of investment might be
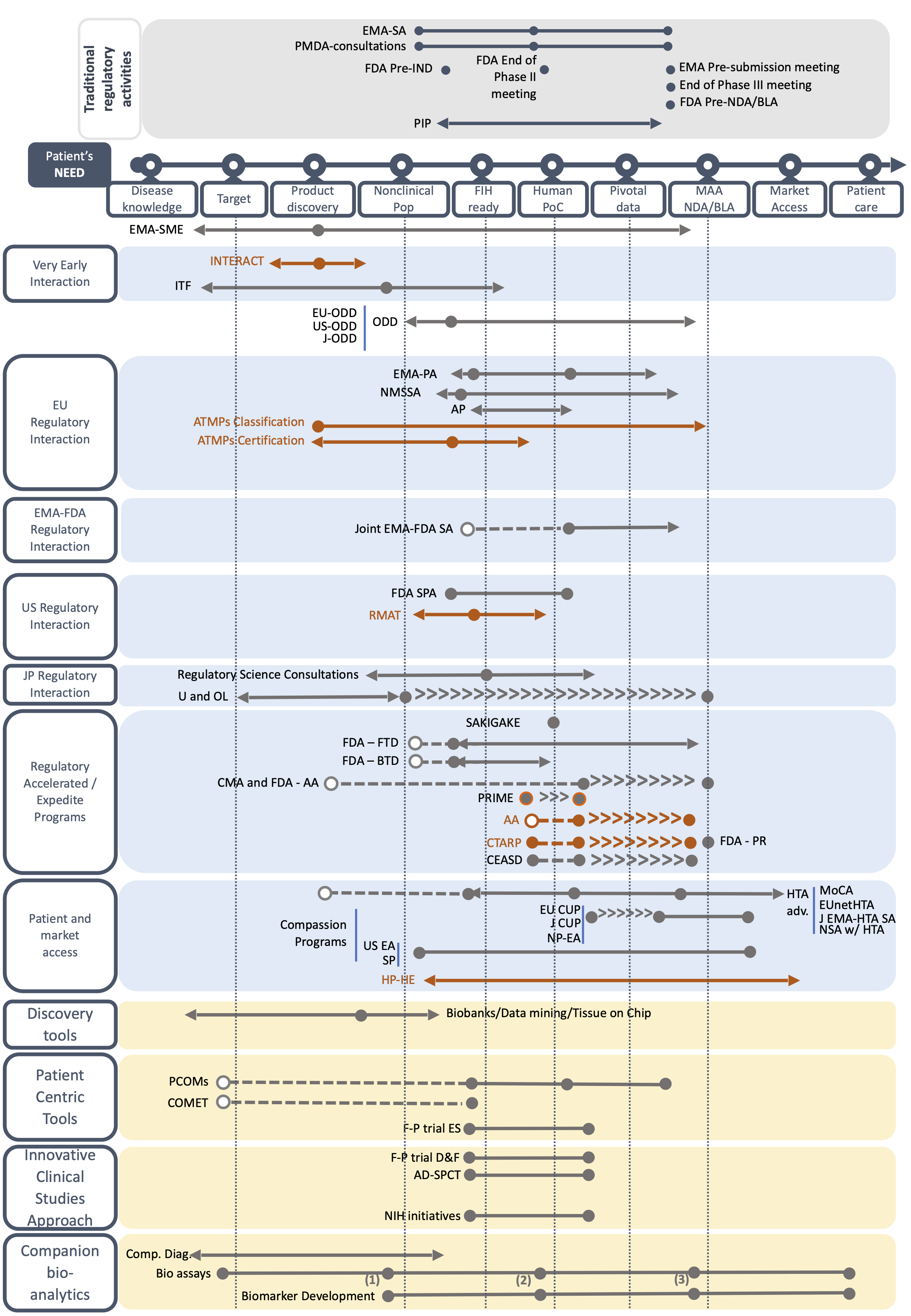
TRADITIONAL TECHNOLOGY TARGETING A 'LESS UNDERSTOOD' RARE DISEASE
Case of development of a traditional, well-understood pharmaceutical (such as a small molecule or a protein) targeting a 'less understood' rare disease [i.e., extremely rare, extremely low prevalence, neglected (i.e., complete unmet medical need), scattered diseases knowledge, and no recognized endpoints or biomarkers, pediatric only indication] to be registered in EU, US and Japan
-
Assumptions
- The drug is a well-understood pharmaceutical product
- Prevalence of the disease is < 1 per-million inhabitants in all geographies
- Medical body of knowledge around the disease is negligible
- Natural history of the disease is little known; no severity or prognostic phenotypes have been identified so far
- The disease exists only in pediatric patients
- No biomarkers or clinically-relevant endpoints are available
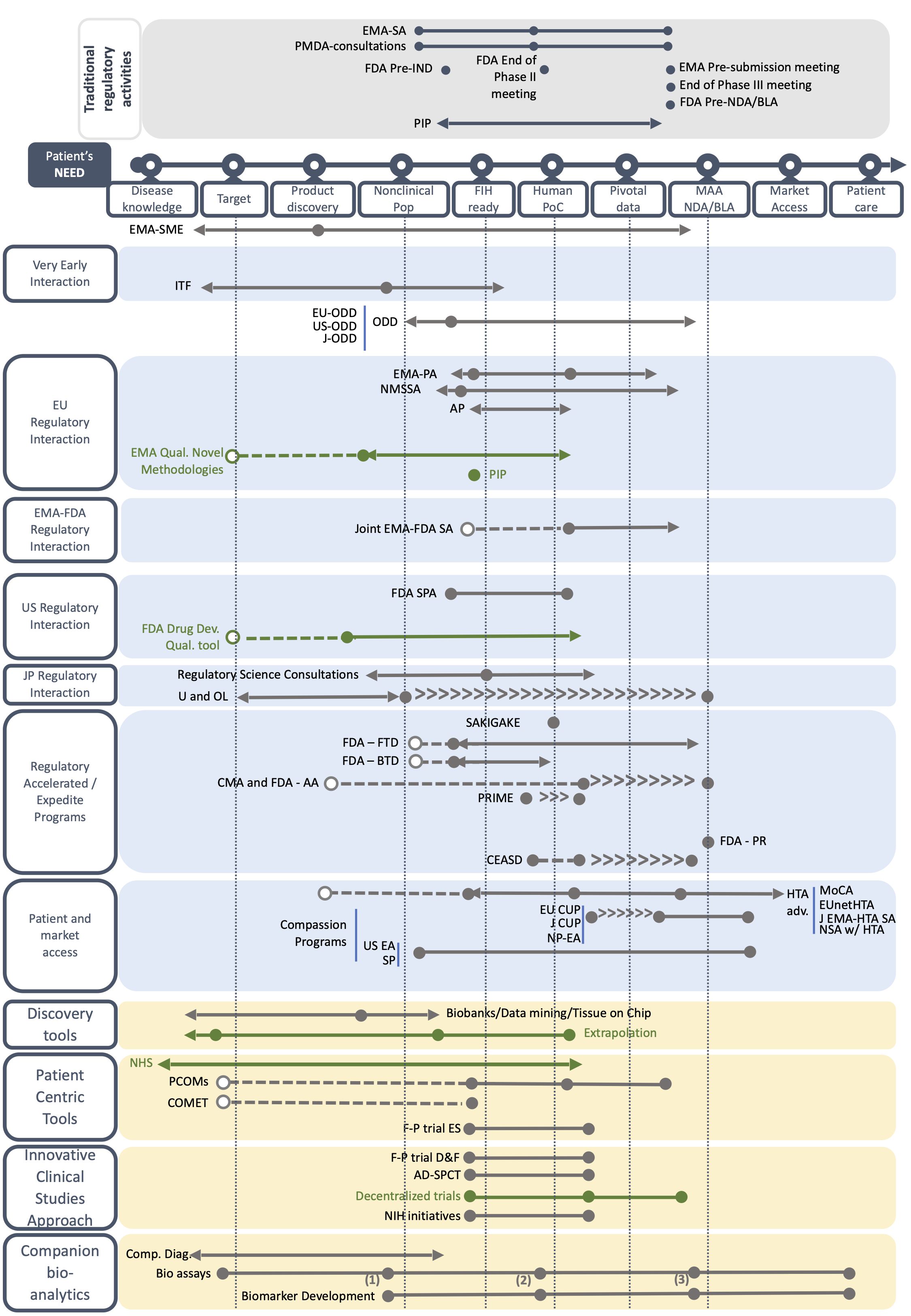
HIGHLY INNOVATIVE TECHNOLOGY TARGETING A 'LESS UNDERSTOOD' RARE DISEASE
Case of development of a highly innovative technology (i.e. advanced therapy medicinal products) targeting a 'less understood' rare disease (i.e., extremely low prevalence, scattered diseases knowledge, and no recognized endpoints or biomarkers)
-
Assumptions
- The drug is an highly innovative technology like the advanced therapy medicinal products
- Prevalence of the disease is < 1 per-million inhabitants in all geographies
- Medical body of knowledge around the disease is negligible
- Natural history of the disease is little known; no severity or prognostic phenotypes have been identified so far
- The disease exists only in pediatric patients
- No biomarkers or clinically-relevant endpoints are available
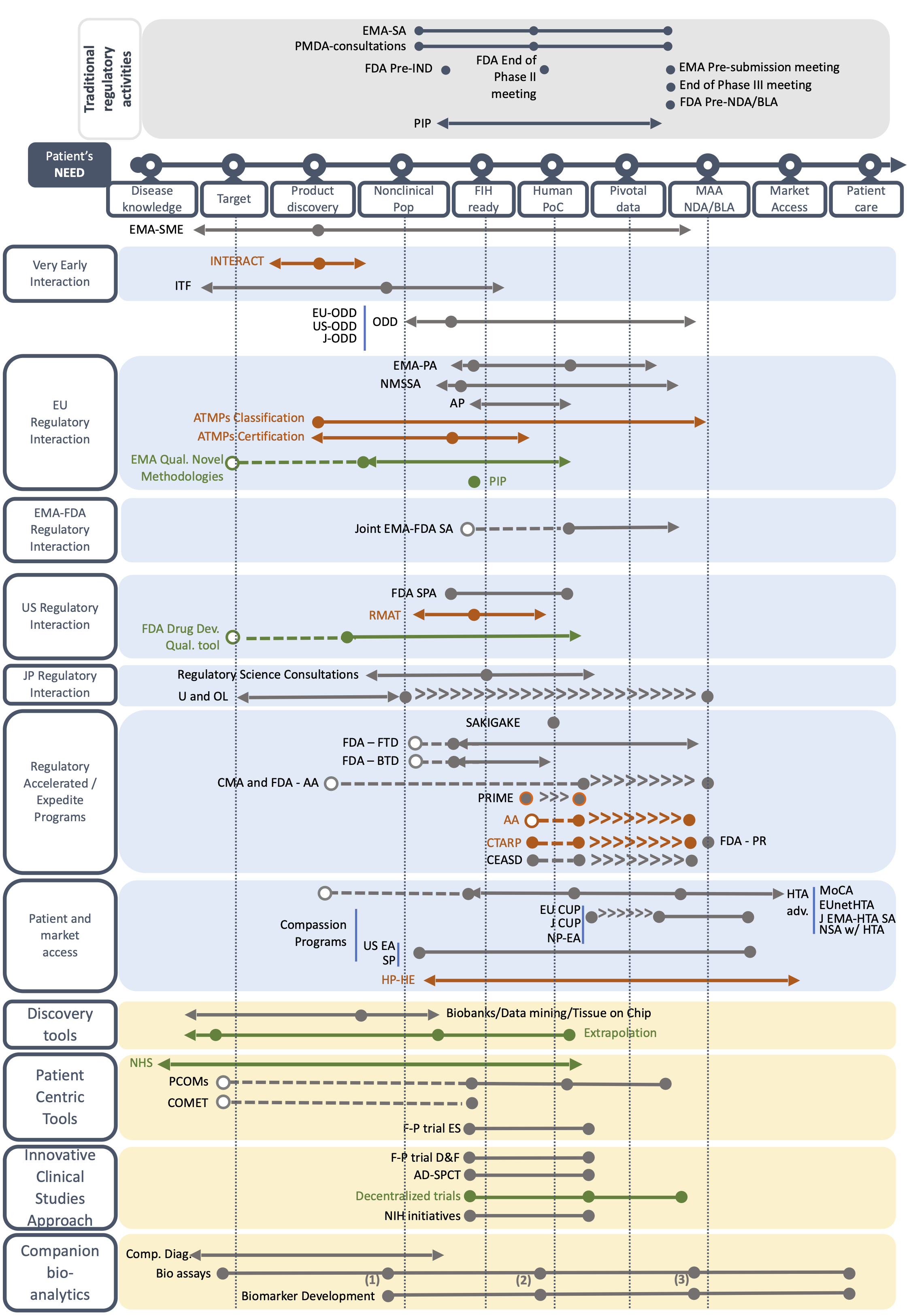
Drug Repurposing Development Cases
DRUG REPURPOSING CASE FOR A SUFFICIENTLY WELL UNDERSTOOD DISEASE
Repurposing of an active substance that is well-understood (a small molecule) targeting a 'sufficiently well understood' rare disease to be registered for the new indication in EU, US and Japan
-
Assumptions
- The active substance is well-understood, and has already been registered for a previous indication, as part of a drug product with specific dose strength
- The active substance that will be repurposed no longer is on patent
- The developer doing the repurposing is not the marketing authorization holder
- The active substance to be repurposed will be used as a monotherapy
- The repurposed pharmaceutical product makes use of the same mechanism of action
- There is already a non-negligible body of knowledge around the disease in the regulatory and medical community
- Patient population is sufficiently understood
- The repurposed pharmaceutical product is given in the same pharmaceutical form as the drug for the original indication
- Funding the development is not a problem, but return on investment might be
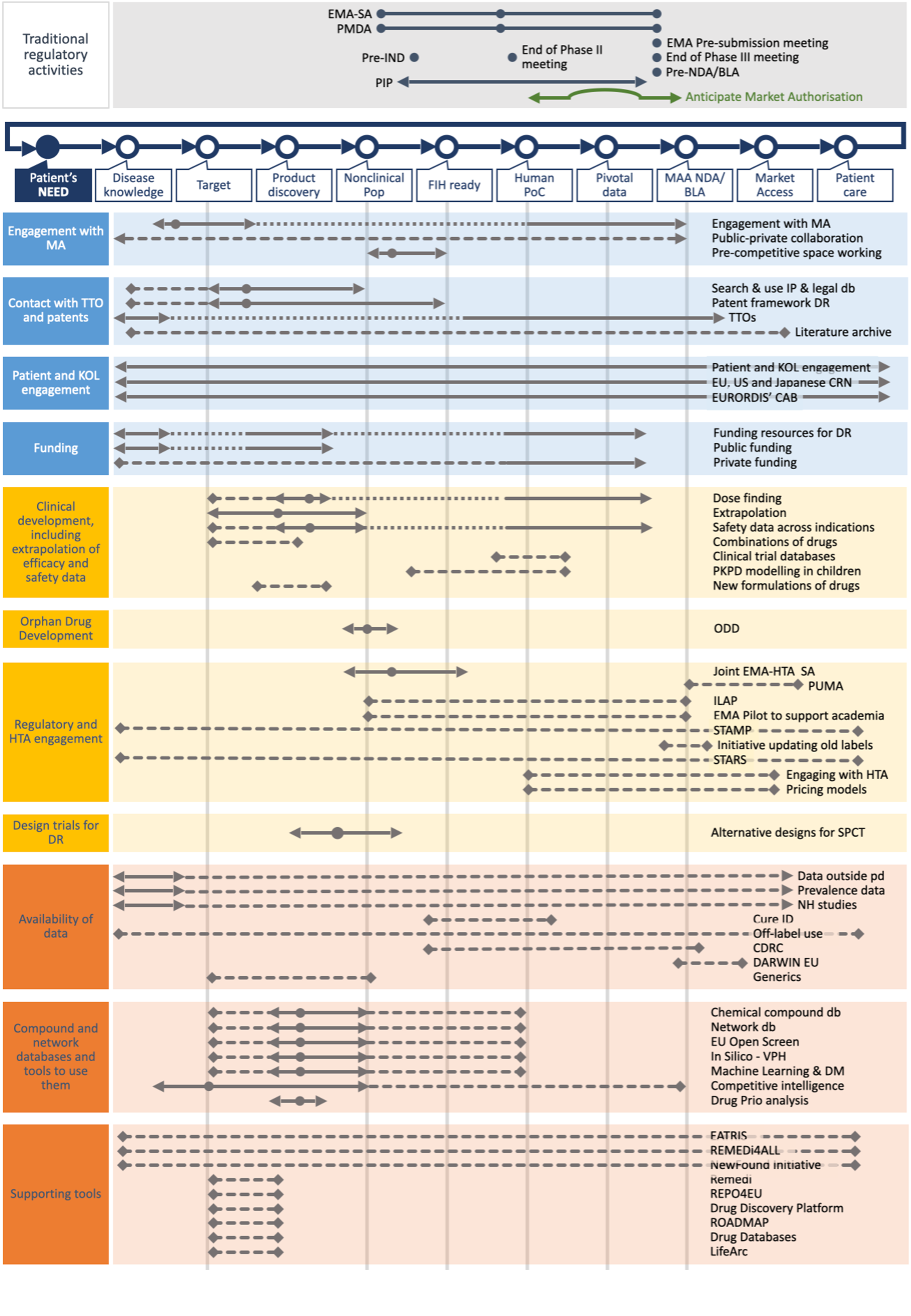
Building Blocks
Based on a systematic review of websites, literature search and the expertise of the Taskforce members, we have created fact sheets describing each BB by including key information on its use, duration, pros and cons, and among other aspects the TF’s advice on the best time to use the BB. All fact sheets can be found below. You can access to the BB forms by type or by geographical scope, or by type of development (i.e. orphan drug development or drug repurposing).
E101 - EMA Innovation Task Force [ITF]
Europe Regulatory Orphan DrugE102 - European Orphan Drug Designation [EU-ODD]
Europe Regulatory Orphan DrugE103 - EMA Protocol Assistance (i.e., Scietific Advice for Orphans) [EMA-PA or EMA-SA]
Europe Regulatory Orphan DrugE104 - National Member State Scientific Advice [NMSSA]
Europe Regulatory Orphan DrugE105 - European Medicines Agency's Scientific Advisory Groups [SAGs]
Europe Regulatory Orphan DrugE106 - EMA Priority Medicines -PRIME scheme [PRIME]
Europe Regulatory Orphan DrugE107 - Accelerated assessment [AA]
Europe Regulatory Orphan DrugE108 - Conditional Marketing Authorization [CMA]
Europe Regulatory Orphan DrugE109 - Marketing Authorization under Exceptional circumstances [MAUEC]
Europe Regulatory Orphan DrugE110 - EMA Qualification of novel methodologies for medicine development [EMA-Qual.Novel Methodologies]
Europe Regulatory Orphan DrugE111 - Potential for Synergy with Pediatric Regulation (+2 years of Market exclusivity)
Europe Regulatory Orphan DrugE112 - Paediatric Investigation Plans [PIP]
Europe Regulatory Orphan DrugE113 - Adaptive Pathways [AP]
Europe Regulatory Orphan DrugE114 - Certification for advanced therapy medicinal products [ATMPs certification]
Europe Regulatory Orphan DrugE115 - EMA small-medium entreprizes (SME) office [SME]
Europe Regulatory Orphan DrugE116 - EMA Framework for stakeholder’s engagement – patients, academia, healthcare professionals
Europe Regulatory Orphan DrugE117 - Business Pipeline Meetings
Europe Regulatory Orphan DrugE118 - EMA Marketing Authorization (MA) with conditions [MA]
Europe Regulatory Orphan DrugE119 - EMA Initiative for Patient Registries
Europe Regulatory Orphan DrugE120 - Mechanism of Coordinated Access to OMPs [MoCA]
Europe HTA and reimbursement Orphan DrugE121 - European Network for Health Technology Assessment [EUNetHTA]
Europe HTA and reimbursement Orphan DrugE122 - EU Compassionate Use Programs [EU CUP]
Europe Early access Orphan DrugE123 - European Commission funded programs and resources
Europe Development resources Orphan DrugE124 - European Joint Programme for Rare Diseases [EJP-RD]
Europe Development resources Orphan DrugE125 - European Reference Networks [ERNs]
Europe Development resources Orphan DrugE126 - EURORDIS’ Community Advisory Boards [CABs]
Europe Development resources Orphan DrugE127 - European Patients' Academy (EUPATI) toolbox [EUPATI]
Europe Development resources Orphan DrugE128 - Connect 4 Children (C4C) – Pediatric Clinical Research Networks [C4C]
Europe Development resources Orphan DrugE129 - EURORDIS Open Academy
Europe Development resources Orphan DrugE130 - European Network of Paediatric Research at the European Medicines Agency [EnprEMA]
Europe Development resources Orphan DrugE131 - Core Outcome Measures in Effectiveness Trials [COMET]
Europe Development resources Orphan DrugE132 - EU planned cross-border treatments
Europe Early access Orphan DrugE133 - Advanced Therapy Medicinal Products (ATMPs) Classification [ATMPs classification]
Europe Regulatory Orphan DrugE134 - Joint EMA-HTA Scientific Advice [J EMA-HTA SA]
Europe Regulatory Orphan DrugE135 - Magisterial hospital preparations – hospital exemptions [HP-HE]
Europe Early access Orphan DrugE136 - EMA pre-submission meeting (Pre-MAA meetings) [EMA pre-submission meeting (Pre-MAA meetings)]
Europe Regulatory Orphan DrugI401 - Joint EMA-FDA Scientific Advice [Joint EMA-FDA SA]
International Regulatory Orphan DrugI402 - Extrapolation of efficacy and safety in medicine development [Extrapolation]
International Development practices Orphan DrugI403 - National Scientific Advice with HTA bodies [NSA w/ HTA]
International HTA and reimbursement Orphan DrugI404 - Crowd funding
International HTA and reimbursement Orphan DrugI405 - National programs for early access [NP-EA]
International Early access Orphan DrugI406 - Tissue Chip for Drug Screening program and Consortium [Tissue Chip]
International Development resources Orphan DrugI407 - Data mining [Data mining]
International Development practices Orphan DrugI408 - BBMRI-ERIC - European research infrastructure for biobanking. [Biobanks]
International Development resources Orphan DrugI409 - Orphanet database
International Development resources Orphan DrugI410 - NCATS and NHGRI Genetic and Rare Diseases Information Center [GARD]
International Development resources Orphan DrugI411 - Coding of Rare Diseases: Orphanet nomenclature
International Development resources Orphan DrugI412 - Decentralized trials [Decentralized trials ]
International Development practices Orphan DrugI413 - Use of Biomarkers in Orphan Drug Development [Biomarkers Development]
International Development practices Orphan DrugI414 - Patient surveys / Patient Preferences studies / Ethnographic research
International Development practices Orphan DrugI415 - Development and use of Patient-Centered Outcome Measures [PCOM]
International Development practices Orphan DrugI416 - Compagnion diagnostics [Comp. Diag.]
International Development practices Orphan DrugI417 - Development of product specific bio-analytical assays [Bio-assays]
International Development practices Orphan DrugI418 - Natural History Studies (NHS) [NHS]
International Development practices Orphan DrugI419 - Registries for Rare Diseases
International Development practices Orphan DrugI420 - Private funding
International Development resources Orphan DrugI421 - Alternative designs for Small Population Clinical Trials [AD-SPCT]
International Development practices Orphan DrugI422 - Feasibility-Patient engagement in trial design and feasibility [F-P trial D&F]
International Development practices Orphan DrugI423 - Feasibility-Patient engagement in trial endpoint selection [F-P trial ES]
International Development practices Orphan DrugI424 - Ethic's Committees assessment of protocols for trials with orphan medications
International Development practices Orphan DrugI425 - FAIR principle for data use
International Development practices Orphan DrugI426 - Initiatives for undiagnosed diseases
International Development resources Orphan DrugI427 - New-born screening programs
International Development resources Orphan DrugI428 - IRDiRC Recognized Resources
International Development resources Orphan DrugI429 - TREAT-NMD Advisory Committee for Therapeutics [TACT]
International Development resources Orphan DrugI430 - Technology transfer offices [TTOs]
International Development resources Orphan DrugI431 - Target Patient Value Profile
International Development practices Orphan DrugI432 - Horizon Scanning: Landscape analysis/ Stakeholder identification and engagement
International Development practices Orphan DrugJ301 - Japanese Orphan Drugs/Medical Devices/Regenerative Medical Products Designation [J-ODD]
Japan Regulatory Orphan DrugJ302 - Sakigake Designation System [SAKIGAKE ]
Japan Regulatory Orphan DrugJ303 - PMDA consultations [PMDA]
Japan Regulatory Orphan DrugJ304 - Regulatory Science General Consultation and Regulatory Science Strategy Consultation (R&D) [Regulatory Science Consultations]
Japan Regulatory Orphan DrugJ305 - Consultations on Pharmacogenomics / Biomarkers (As part of PMDA consultations)
Japan Regulatory Orphan DrugJ306 - Conditional Early Approval System for Drugs
Japan Regulatory Orphan DrugJ307 - Conditional Early Approval System for Medical Devices [CEASD]
Japan Regulatory Orphan DrugJ308 - Conditional and Time-limited Authorization of Regenerative Medical Products [CTARP]
Japan Regulatory Orphan DrugJ309 - Designated Intractable Diseases (Designated Nan-byo)
Japan Regulatory Orphan DrugJ310 - Study Group on Unapproved and Off-label Drugs of High Medical Need [U and OL]
Japan Regulatory Orphan DrugJ311 - Japan Compassionate Use Programs [J CUP]
Japan Early access Orphan DrugJ312 - AMED funded programs and resources
Japan Development resources Orphan DrugJ313 - AMED – Initiative on Rare and Undiagnosed Diseases [IRUD]
Japan Development resources Orphan DrugJ314 - AMED Clinical Innovation network – Diseases-related Registries
Japan Development resources Orphan DrugU201 - US Orphan Drug Designation [US-ODD]
USA Regulatory Orphan DrugU202 - Humanitarian Device
USA Regulatory Orphan DrugU203 - FDA Expedited Program for serious conditions - Fast Track Designation [FDA-FTD]
USA Regulatory Orphan DrugU204 - FDA Expedited Program for serious conditions - Breakthrough Therapy Designation [FDA-BTD]
USA Regulatory Orphan DrugU205 - FDA Expedited Program for serious conditions - Accelerated Approval [FDA-AA]
USA Regulatory Orphan DrugU206 - FDA Expedited Program for serious conditions - Priority Review Designation [FDA-PR]
USA Regulatory Orphan DrugU207 - FDA Expedited Program for serious conditions - Special Protocol Assessment [FDA-SPA]
USA Regulatory Orphan DrugU208 - Rare Pediatric Disease Designation
USA Regulatory Orphan DrugU209 - Rare Pediatric Priority Review Voucher
USA Regulatory Orphan DrugU210 - Neglected and Tropical Diseases Priority Review Voucher
USA Regulatory Orphan DrugU211 - Regenerative Medicine Advanced Therapy (RMAT) Designation [RMAT]
USA Regulatory Orphan DrugU212 - FDA Milestones meetings – Type B [FDA End of Phase II meeting/ End of Phase III meeting / FDA Pre-NDA/BLA]
USA Regulatory Orphan DrugU213 - FDA Type C, A meetings
USA Regulatory Orphan DrugU214 - Pre Investigational New Drug (IND) interactions [FDA Pre-IND]
USA Regulatory Orphan DrugU215 - Product specific- pre approval advisory committee
USA Regulatory Orphan DrugU216 - FDA Pediatrics Advisory Committee
USA Regulatory Orphan DrugU217 - Stakeholders interactions - Professional affairs and Community Engagement
USA Regulatory Orphan DrugU218 - Stakeholders interactions - Patients Affairs Staff [PAS]
USA Regulatory Orphan DrugU219 - FDA Qualification process - Drug Development Tools [FDA Drug Dev. Qual. Tool]
USA Regulatory Orphan DrugU220 - Right to Try Act [RtT]
USA Regulatory Orphan DrugU221 - Patient-focused drug development program
USA Regulatory Orphan DrugU222 - INTERACT Meetings (Initial Targeted Engagement for Regulatory Advice on CBER products) [INTERACT]
USA Regulatory Orphan DrugU223 - NCAT Toolkit for Patients-Focused Therapy Development
USA Regulatory Orphan DrugU224 - US Expanded Access Program [US EA]
USA Early access Orphan DrugU225 - Single Patient Expanded Access [SP]
USA Early access Orphan DrugU226 - NIH funded programs and resources [NIH initiatives]
USA Development resources Orphan DrugU227 - Clinical Research Networks [CRN]
USA Development resources Orphan DrugU228 - Discovering New Therapeutics Uses for Existing Molecules (NTU) program, National Center for Advancing Translational Sciences (NCATS) [NTU-NCATS]
USA Development resources Orphan DrugI433 - Cure ID [C-ID]
International Development practices RepurposingI434 - Off label use [OLU]
International Development practices RepurposingI435 - Cure Drug Repurposing Collaboratory [CDRC]
International Development resources RepurposingE147 - DARWIN EU [D-EU]
Europe Regulatory RepurposingI436 - Chemical compound databases [CCDB]
International Development resources RepurposingI437 - Network databases [NDB]
International Development resources RepurposingE137 - PUMA [PUMA]
Europe Regulatory RepurposingE138 - ILAP [ILAP]
Europe Regulatory RepurposingE139 - EMA pilot project to support academia [ESA]
Europe Regulatory RepurposingE140 - STAMP initiative [STAMP]
Europe Regulatory RepurposingU229 - Initiative updating old labels - BPCA + PREA [IOL]
United States Regulatory RepurposingE141 - STARS [STARS]
Europe Regulatory RepurposingI438 - Engaging with MA holders [EMAH]
International Development practices RepurposingE142 - EATRIS [EATRIS]
Europe Development resources RepurposingI439 - NewFound Initiative [NFI]
International Development resources RepurposingI440 - In silico models for screening for drug repurposing candidates [VPH]
International Development practices RepurposingI441 - Machine learning & data mining [ML&DM]
International Development practices RepurposingI442 - Competitive intelligence [CI]
International Development practices RepurposingI443 - Drug prioritization analysis [DPA]
International Development practices RepurposingI444 - How to search and use IP and legal databases [IP&L]
International Development practices RepurposingI445 - Patent Framework of Drug Repurposing [PF-DR]
International Development resources RepurposingI446 - Regulatory Framework of Drug Repurposing [RF-DR]
International Development practices RepurposingI447 - How to maintain a literature archive [MLA]
International Development practices RepurposingI448 - Funding resources for drug repurposing [FR]
International Development resources RepurposingE143 - EU openscreen [EU-O]
Europe Development resources RepurposingI449 - REPO4EU [REPO4EU]
International Development resources RepurposingE144 - REMEDI4All [REMEDI4ALL]
Europe Development resources RepurposingI450 - Drug discovery platform [DDP]
International Development resources RepurposingU230 - ROADMAP [ROADMAP]
United States Development resources RepurposingI451 - Remedi [RE]
International Development resources RepurposingI452 - Generics [GE]
International Development resources RepurposingI453 - Drug databases [DDB]
International Development resources RepurposingI454 - Combination of drugs [C-DR]
International Development practices RepurposingI455 - Clinical trials database [CT-DB]
International Development resources RepurposingI456 - PKPD Modelling in children [PKPDM]
International Development practices RepurposingI457 - Dose finding [DF]
International Development practices RepurposingI458 - new formulation of drugs [NFD]
International Development practices RepurposingI459 - safety data across indications [SDAI]
International Development resources RepurposingI460 - Data outside the public domain [DO-PD]
International Development resources RepurposingI461 - How to develop pricing models for repurposing [PR-Models]
International HTA and reimbursement RepurposingE145 - LifeArc Repurposing Medicines Toolkit [LA]
Europe Development resources RepurposingI462 - Public-private partnerships [PPR]
International Development practices RepurposingI463 - Pre-competitive space working [PCSW]
International Development practices RepurposingU231 - Updating old labels - Project Renewal [RENEWAL]
United States Regulatory RepurposingE146 - Engaging with HTA [E-HTA]
Europe HTA and reimbursement RepurposingU232 - Updating old labels - SRLC [SRLC]
United States Regulatory RepurposingMilestones
For both orphan drug development and drug repurposing, several milestones are used throughout the development. These milestones include Target and Product Milestone; Nonclinical POP milestone; First-in-human ready milestone; Human POC milestone; Pivotal Data milestone; and MAA- NDA/BLA milestone. Please note that there is no First-in-human milestone for drug repurposing.
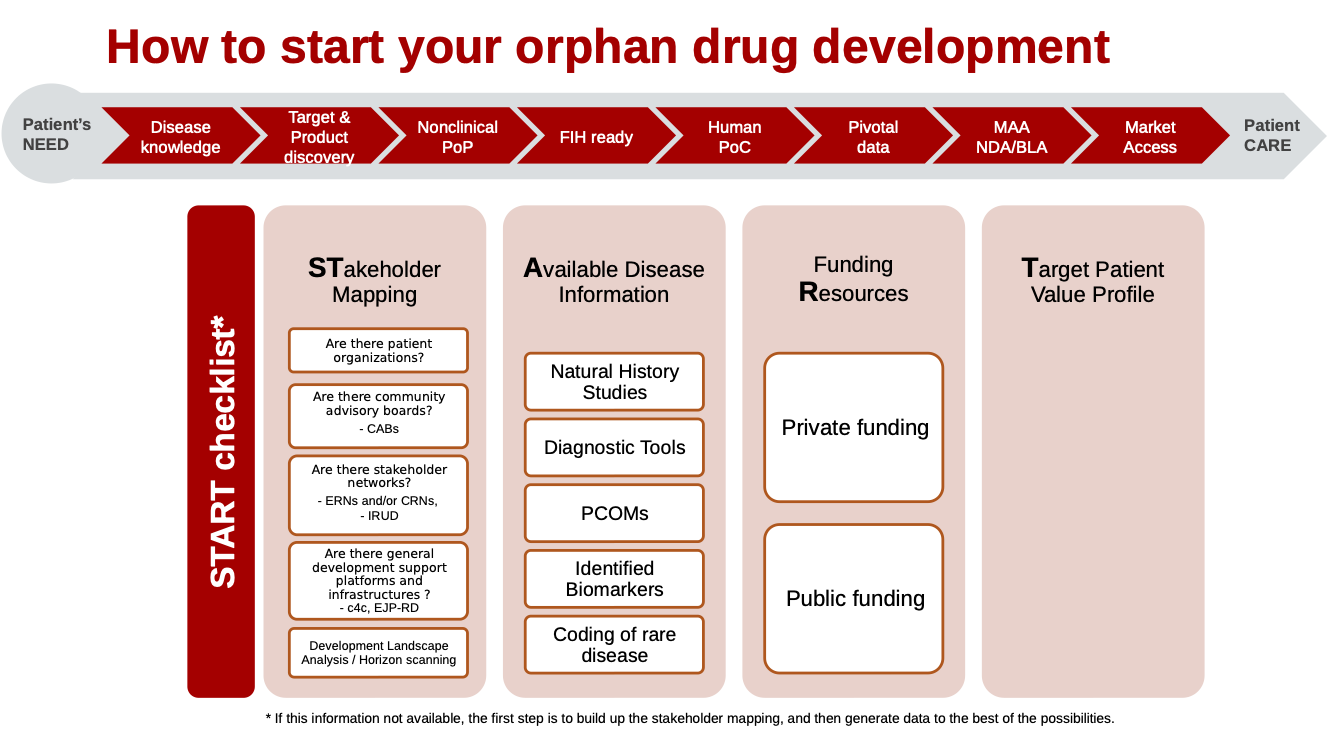
Checklists
There are different checklists available for each milestone, to assure that all the steps are validated in order to move forward. While all checklists are relevant for the milestones of orphan drug development and repurposing (except the first-in-human checklist), a specific checklist for repurposing has been developed, and is included at the bottom of the page. Please note that there is no First-in-human milestone for drug repurposing, as the drug is repurposed and therefore already tested in humans.
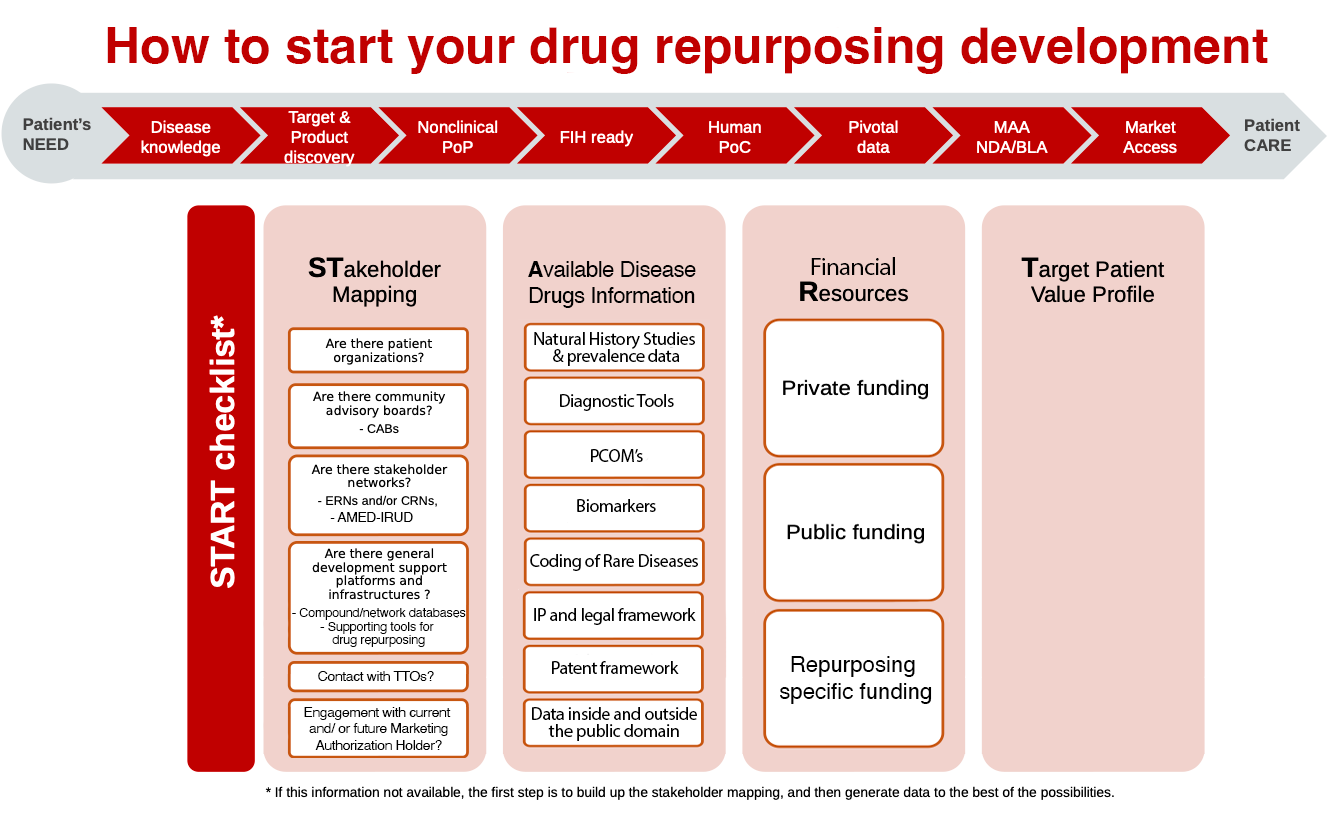
Drug Repurposing Development Checklist Drug Repurposing
-
Description of Drug Repurposing Development Checklist
A checklist with questions that can assist you in your repurposing drug development program
Target and Product Discovery Milestone before
-
Description of Target and Product Discovery Milestone
Typically, researchers discover new drugs through:
- New insights into a disease process that allow researchers to design a product to stop or reverse the effects of the disease.
- Many tests of molecular compounds to find possible beneficial effects against any of a large number of diseases.
- Existing treatments that have unanticipated effects.
- New technologies, such as those that provide new ways to target medical products to specific sites within the body or to manipulate genetic material.
To select the best drug, researchers engage in an often long phase of work to deepen the understanding of the molecular/ cellular mechanisms to be targeted and its impact on the disease processes, as well as building virtual, cellular, and animal models that reliably reproduce the role of such mechanism. This phase is called Target Validation and enables researchers and developers to create the basic knowledge and tools that will be needed inn the following phases. At this stage in the process, thousands of compounds may be potential candidates for development as a medical treatment. After early testing, however, only a small number of compounds look promising and call for further study. Once researchers identify a promising compound for development, they conduct experiments to gather information on:
- How it is absorbed, distributed, metabolized, and excreted.
- Its potential benefits and mechanisms of action.
- The best dosage.
- The best way to give the drug (such as by mouth or injection).
- Side effects or adverse events that can often be referred to as toxicity.
- How it affects different groups of people (such as by gender, race, or ethnicity) differently.
- How it interacts with other drugs and treatments.
- Its effectiveness as compared with similar drugs.
This fishing and selection phase is called Drug Discovery: its scope is to produce a single (or limited number) of best candidate products responding to all the characteristics that can make them (in theory and experimentally in the lab) a suitable drug for patients.
Target and Product Discovery Milestone after
-
Description of Target and Product Discovery Milestone
Typically, researchers discover new drugs through:
- New insights into a disease process that allow researchers to design a product to stop or reverse the effects of the disease.
- Many tests of molecular compounds to find possible beneficial effects against any of a large number of diseases.
- Existing treatments that have unanticipated effects.
- New technologies, such as those that provide new ways to target medical products to specific sites within the body or to manipulate genetic material.
To select the best drug, researchers engage in an often long phase of work to deepen the understanding of the molecular/ cellular mechanisms to be targeted and its impact on the disease processes, as well as building virtual, cellular, and animal models that reliably reproduce the role of such mechanism. This phase is called Target Validation and enables researchers and developers to create the basic knowledge and tools that will be needed inn the following phases. At this stage in the process, thousands of compounds may be potential candidates for development as a medical treatment. After early testing, however, only a small number of compounds look promising and call for further study. Once researchers identify a promising compound for development, they conduct experiments to gather information on:
- How it is absorbed, distributed, metabolized, and excreted.
- Its potential benefits and mechanisms of action.
- The best dosage.
- The best way to give the drug (such as by mouth or injection).
- Side effects or adverse events that can often be referred to as toxicity.
- How it affects different groups of people (such as by gender, race, or ethnicity) differently.
- How it interacts with other drugs and treatments.
- Its effectiveness as compared with similar drugs.
This fishing and selection phase is called Drug Discovery: its scope is to produce a single (or limited number) of best candidate products responding to all the characteristics that can make them (in theory and experimentally in the lab) a suitable drug for patients.
Nonclinical PoP milestone before
-
Description of Nonclinical PoP milestone
PoP: Proof of Principle Pre-clinical research is a preliminary phase that involves testing the drug in lab models (in-vitro) and on animals, including basic testing for safety flags. Before testing a drug in humans, researchers must find out whether it has the potential to cause serious harm, also called toxicity. The two types of preclinical research are:
- In vitro (i.e. by chemical testing, in cells, in organoids or in isolated tissues or organs)
- In vivo (i.e. in animals)
PoP is achieved when a sufficient evidence of biological activity in vitro and in vivo is demonstrated, an adequate understanding of the pharmaco-kinetics and pharmaco-dynamics of the drug is gathered, and some initial toxicology and safety information is generated. With these data, researchers can plan the following phase of extensive testing of toxicology and biodistribution, which is required to move into clinical studies.
Nonclinical PoP milestone after
-
Description of Nonclinical PoP milestone
PoP: Proof of Principle Pre-clinical research is a preliminary phase that involves testing the drug in lab models (in-vitro) and on animals, including basic testing for safety flags. Before testing a drug in humans, researchers must find out whether it has the potential to cause serious harm, also called toxicity. The two types of preclinical research are:
- In vitro (i.e. by chemical testing, in cells, in organoids or in isolated tissues or organs)
- In vivo (i.e. in animals)
PoP is achieved when a sufficient evidence of biological activity in vitro and in vivo is demonstrated, an adequate understanding of the pharmaco-kinetics and pharmaco-dynamics of the drug is gathered, and some initial toxicology and safety information is generated. With these data, researchers can plan the following phase of extensive testing of toxicology and biodistribution, which is required to move into clinical studies.
First-in-Human ready milestone before
-
Description of First-in-Human ready milestone
Purpose: Safety and dosage In order to enable starting human testing of pharmaceuticals, Regulatory Authorities require researchers to test product’s safety in animals (in vivo toxicology) at doses higher than the corresponding future human doses in highly controlled experiments. the Good Laboratory Practices (GLP) for Nonclinical Laboratory Studies set the minimum basic requirements for conducting “clinical study enabling” toxicology experiments regarding:
- study conduct
- personnel
- facilities
- equipment
- written protocols
- operating procedures
- study reports
- and a system of quality assurance oversight for each study to help assure the safety of the product
Usually, preclinical studies are not very large. However, these studies must provide detailed information on dosing and toxicity levels. After preclinical testing, researchers review their findings and decide whether the drug should be tested in humans.
First-in-Human ready milestone after
-
Description of First-in-Human ready milestone
Purpose: Safety and dosage In order to enable starting human testing of pharmaceuticals, Regulatory Authorities require researchers to test product’s safety in animals (in vivo toxicology) at doses higher than the corresponding future human doses in highly controlled experiments. the Good Laboratory Practices (GLP) for Nonclinical Laboratory Studies set the minimum basic requirements for conducting “clinical study enabling” toxicology experiments regarding:
- study conduct
- personnel
- facilities
- equipment
- written protocols
- operating procedures
- study reports
- and a system of quality assurance oversight for each study to help assure the safety of the product
Usually, preclinical studies are not very large. However, these studies must provide detailed information on dosing and toxicity levels. After preclinical testing, researchers review their findings and decide whether the drug should be tested in humans.
Human PoC milestone before
-
Description of Human PoC milestone
PoC: Proof of Concept Purpose: Efficacy and side effects While preclinical research answers basic questions about a drug’s safety, it is not a substitute for studies of ways the drug will interact with the human body. “Clinical research” refers to studies, or trials, that are done in people. As the developers design the clinical study, they will consider what they want to accomplish for each of the different Clinical Research Phases and begin the Investigational New Drug Process (IND) / Clinical Trial Application (CTA) / national regulatory applications, a process they must go through before clinical research begins. The First in Human (FIH) is the first clinical trial where the drug tested previously in animal is for the first time tested in normal volunteers (healthy people). In most cases, 20 to 80 healthy volunteers or people with the disease/condition participate in this first study (or set of studies) aiming at providing initial safety and tolerability information. This set of studies in healthy volunteers is collectively called “phase I”. However, if a new drug is intended for use in cancer patients or if the administration of the drug poses risks to the healthy volunteers (e.g. in the case of most biotechnology products), researchers conduct phase I studies in patients with that type of cancer/rare-disease/etc. Phase I studies are closely monitored and gather information about how a drug interacts with the human body. Researchers adjust dosing schemes based on animal data to find out how much of a drug the body can tolerate and what its acute side effects are. As a phase I trials continue, researchers answer research questions related to how it works in the body, the side effects associated with increased dosage, and early information about how effective it is to determine how best to administer the drug to limit risks and maximize possible benefits. This is important to the design of phase II studies. In some cases, the phase I and II may be condensed.
- Study Participants (Phase I): 20 to 100 healthy volunteers or people with the disease/condition.
- Length of Study: Several months
- Purpose: Safety and dosage
In phase II studies, researchers administer the drug to a (small) group of patients with the disease or condition for which the drug is being developed. The scope of phase II studies is to provide the first evidence of biological activity, efficacy and safety in the intended patient population, as well as to selected the best dose(s) to be further studied in phase III. Typically involving a few hundred patients, these studies aren't large enough to formally demonstrate whether the drug will be beneficial, or to accurately predict product’s safety. In rare diseases, phase II (or combined phase I-II) studies might be much smaller, sometimes only a few dozens of patients or even less. Researchers use data gathered in phase II to refine research questions, develop research methods, and design phase III research protocols.
- Study Participants (Phase II): Up to several hundred people with the disease/condition.
- Length of Study: Several months to 2 years
- Purpose: Efficacy and side effects
Human PoC milestone after
-
Description of Human PoC milestone
PoC: Proof of Concept Purpose: Efficacy and side effects While preclinical research answers basic questions about a drug’s safety, it is not a substitute for studies of ways the drug will interact with the human body. “Clinical research” refers to studies, or trials, that are done in people. As the developers design the clinical study, they will consider what they want to accomplish for each of the different Clinical Research Phases and begin the Investigational New Drug Process (IND) / Clinical Trial Application (CTA) / national regulatory applications, a process they must go through before clinical research begins. The First in Human (FIH) is the first clinical trial where the drug tested previously in animal is for the first time tested in normal volunteers (healthy people). In most cases, 20 to 80 healthy volunteers or people with the disease/condition participate in this first study (or set of studies) aiming at providing initial safety and tolerability information. This set of studies in healthy volunteers is collectively called “phase I”. However, if a new drug is intended for use in cancer patients or if the administration of the drug poses risks to the healthy volunteers (e.g. in the case of most biotechnology products), researchers conduct phase I studies in patients with that type of cancer/rare-disease/etc. Phase I studies are closely monitored and gather information about how a drug interacts with the human body. Researchers adjust dosing schemes based on animal data to find out how much of a drug the body can tolerate and what its acute side effects are. As a phase I trials continue, researchers answer research questions related to how it works in the body, the side effects associated with increased dosage, and early information about how effective it is to determine how best to administer the drug to limit risks and maximize possible benefits. This is important to the design of phase II studies. In some cases, the phase I and II may be condensed.
- Study Participants (Phase I): 20 to 100 healthy volunteers or people with the disease/condition.
- Length of Study: Several months
- Purpose: Safety and dosage
In phase II studies, researchers administer the drug to a (small) group of patients with the disease or condition for which the drug is being developed. The scope of phase II studies is to provide the first evidence of biological activity, efficacy and safety in the intended patient population, as well as to selected the best dose(s) to be further studied in phase III. Typically involving a few hundred patients, these studies aren't large enough to formally demonstrate whether the drug will be beneficial, or to accurately predict product’s safety. In rare diseases, phase II (or combined phase I-II) studies might be much smaller, sometimes only a few dozens of patients or even less. Researchers use data gathered in phase II to refine research questions, develop research methods, and design phase III research protocols.
- Study Participants (Phase II): Up to several hundred people with the disease/condition.
- Length of Study: Several months to 2 years
- Purpose: Efficacy and side effects
Pivotal Data milestone before
-
Description of Pivotal Data milestone
Purpose: Efficacy and monitoring of adverse reactions Researchers design phase III studies to demonstrate whether or not a product offers a treatment benefit to a specific population, i.e. whether the benefit-risk ratio of the drug is positive. Sometimes known as pivotal studies, these studies may involve hundred to thousands of participants for large therapeutic indications and dozens to hundred for rare diseases. Data gathered in phase III studies determine the “label” of the product, i.e. the therapeutic indication, the posology and mode of administration, and the expected efficacy and potential risk as reported in the prescribing information leaflet. Phase III studies provide most of the safety data. In previous studies, it is possible that less common side effects might have gone undetected. Because these studies are larger and longer in duration, the results are more likely to show long-term or rare side effects.
- Study Participants (Phase III): 300 to 3,000 volunteers who have the disease or condition
- Length of Study: 1 to 4 years
- Purpose: Efficacy and monitoring of adverse reactions
Pivotal Data milestone after
-
Description of Pivotal Data milestone
Purpose: Efficacy and monitoring of adverse reactions Researchers design phase III studies to demonstrate whether or not a product offers a treatment benefit to a specific population, i.e. whether the benefit-risk ratio of the drug is positive. Sometimes known as pivotal studies, these studies may involve hundred to thousands of participants for large therapeutic indications and dozens to hundred for rare diseases. Data gathered in phase III studies determine the “label” of the product, i.e. the therapeutic indication, the posology and mode of administration, and the expected efficacy and potential risk as reported in the prescribing information leaflet. Phase III studies provide most of the safety data. In previous studies, it is possible that less common side effects might have gone undetected. Because these studies are larger and longer in duration, the results are more likely to show long-term or rare side effects.
- Study Participants (Phase III): 300 to 3,000 volunteers who have the disease or condition
- Length of Study: 1 to 4 years
- Purpose: Efficacy and monitoring of adverse reactions
MAA – NDA / BLA (registration) milestone before
-
Description of MAA – NDA / BLA (registration) milestone
If a drug developer has evidence from preclinical and clinical (phases I to III) research that a drug is safe and effective for its intended use, the applicant can file an application to market the drug to the relevant Regulatory Authority (i.e., FDA, EMA, MHLW/PMDA, etc). The relevant Regulatory Authority review team thoroughly examines all submitted data on the drug, decides if it is complete and makes a decision to approve or not to approve it. In cases where the Regulatory Authority determines that a drug has been shown to be safe and effective for its intended use, it is then necessary to work with the applicant to develop and refine prescribing information. This is referred to as “labeling.” Labeling accurately and objectively describes the basis for approval and how best to use the drug.
MAA – NDA / BLA (registration) milestone after
-
Description of MAA – NDA / BLA (registration) milestone
If a drug developer has evidence from preclinical and clinical (phases I to III) research that a drug is safe and effective for its intended use, the applicant can file an application to market the drug to the relevant Regulatory Authority (i.e., FDA, EMA, MHLW/PMDA, etc). The relevant Regulatory Authority review team thoroughly examines all submitted data on the drug, decides if it is complete and makes a decision to approve or not to approve it. In cases where the Regulatory Authority determines that a drug has been shown to be safe and effective for its intended use, it is then necessary to work with the applicant to develop and refine prescribing information. This is referred to as “labeling.” Labeling accurately and objectively describes the basis for approval and how best to use the drug.